Diabetes mellitus is suggested to impact about 1 to 2% of the world’s population.1 Central to the clinical condition is a predisposition toward high blood sugar levels, glucose in particular. While the consequences of persistent hyperglycemia are well documented (microvascular disease with atherosclerosis and compromised renal function, retinopathy and neuro-pathy), the exact role carbohydrates play in the development of these debilitating conditions has not always been clear.2,3
Glucose is the principal metabolic sugar in humans. Monosaccharides such as glucose, galactose, fructose and mannose normally exist in equilibrium between two forms: an open-chain or extended “stickman” form and a closed ring (a Haworth projection form). For glucose, galactose and mannose, an open-chain form exposes a reactive aldehyde (or carbonyl) group on carbon atom #1. This is normally reactive under physiological conditions, causing these sugars to be classified as “reducing” sugars. The aldehyde is particularly reactive toward free amino groups such as those occuring on lysine and arginine residues (Figure 1).1,4 When a reducing sugar’s carbonyl group and a free amine interact, they form an aldimine, or Schiff base, which is reversible. This is, however, the first step toward a later irreversible sugar-amino group complex that is generally known as glycation. The ability of a reducing sugar to form a Schiff base is dependent upon the amount that exists in an open-chain form. Only 0.002% of glucose is normally open-chain, versus 0.005%, 0.020% and 0.700% of mannose, galactose and fructose, respectively.5 Thus, glucose spends less time in its reactive carbonyl form than other sugars, making it the “safe” sugar from a glycation-related, teleological point of view. Nevertheless, glucose-mediated glycation by way of Schiff base formation does occur, and is now being recognized as an inevitable, if not pathological, consequence of normal physiology. In Figure 1, the Amadori product is considered the first glycation product. This will undergo spontaneous and/or microenvironmentally-induced breakdown and rearrangement to form stable end products (advanced glycation endproducts; AGEs). The glycation process occurs both within and outside cells by a variety of sugars.3 The exact products formed vary by the sugar involved and the mechanism of rearrangement. Three general types of glycation products are known:
- a fluorescent structure (i.e. pentosidine) that cross-links lysine and arginine
- a non-fluorescent structure (i.e. imidazole) that also cross-links amino acids
- a non-cross-linking adduct that is covalently attached to a lysine or arginine (i.e. carboxymethyl lysine; CML).1
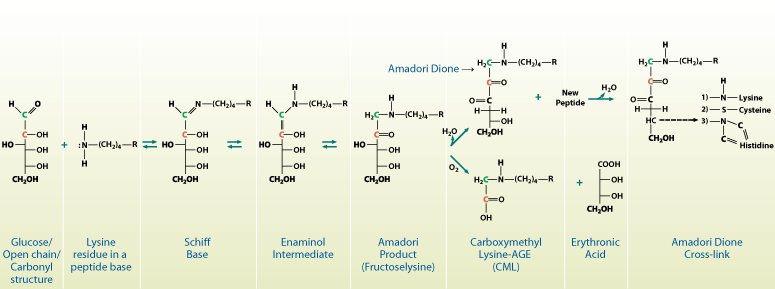 |
| Figure 1. The chemical pathway for the production of glycated proteins. Open-chain glucose reacts with free amines on proteins to form a Schiff base, which is then converted via an enaminol intermediate to the Amadori product or the first glycation product. This product is further modified to produce a cross-link or carboxymethyl lysine (CML). |
The consequences of glycation vary, depending upon the product formed and the molecular structures involved. In general, it could be said that glycation compromises normal molecular structure, function and/or half-life. Add to this the fact that glycated structures are also targets of specific clearance receptors whose ligation promotes inflammation, then one may quickly conclude that glycation has a considerable downside.3,6,7 In a high glucose environment, almost all proteins could be targets for glycation. Four molecules, which are relevant to diabetes, have been studied in some detail, including collagen, albumin, LDL and insulin. Collagen is known to be cross-linked by glucose-derived AGE. This is known to induce an expansion of collagen fibrils, inhibit lateral association of collagen molecules into a normal network-like structure, and promote endothelial cell proliferation and dissociation from basement membrane. This disrupts normal vascular architecture and causes increased vascular permeability.3 Albumin is often glycated, and high levels will promote VEGF expression and neovascularization in the retina,8 vascular smooth muscle proliferation and secretion of inflammatory mediators in the arteries,9 endothelial cell apoptosis with glomerular hyperfiltration in the kidney,10 and an anti-insulin effect in peripheral skeletal muscle.11 Glycation of LDL predisposes it to long-term retention in the tissues, particularly attached to vascular collagen.3,6 Although it has been proposed that retained LDL is more susceptible to oxidation, thus contributing to foam cell formation in vessels, recent data suggests this may not be the case.3,12,13 Finally, insulin, the mediator of glucose influx for most tissues, is also materially glycated in conditions of hyperglycemia.14
Relative to normal glycated insulin levels, diabetic serum can show as much as a two or three-fold increase in glycated insulin.14,15 In experimental models, this could represent as much as 10% of all circulating insulin.14 Glycated insulin will show anywhere from 20 to 40% decreased potency in glucose uptake studies.15 Notably, it appears insulin glycation does not impact insulin-insulin receptor binding, so the effect must be at the post-receptor level.16
References
- Ahmed, N. (2005) Diabetes Res. Clin. Pract. 67:3.
- Singh, R. et al. (2001) Diabetologia 44:129.
- Brownlee, M. (2000) Metabolism 49:9.
- Ulrich, P. & A. Cerami (2001) Recent Prog. Horm. Res. 56:1.
- Bunn, H.F. & P.J. Higgins (1981) Science 213:222.
- Cloos, P.A.C. & S. Christgau (2002) Matrix Biol. 21:39.
- Stern, D.M. et al. (2002) Ageing Res. Rev. 1:1.
- Treins, C. et al. (2001) J. Biol. Chem. 276:43836.
- Hattori, Y. et al. (2002) Hypertension 39:22.
- Amore, A. et al. (2004) Nephrol. Dial. Transplant. 19:53.
- Miele, C. et al. (2003) J. Biol. Chem. 278:47376.
- Bucula, R. et al. (1994) Proc. Natl. Acad. Sci. USA 91:9441.
- Jenkins, A.J. et al. (2004) Metabolism 53:969.
- McKillop. A.M. et al. (2002) Diabetes Metab. 28:3S61.
- Lindsay, J.R. et al. (2003) Diabetologia 46:475.
- Hunter, S.J. et al. (2003) Diabetes 52:492.